Abstract
The interaction between sunlight and drugs can lead to phototoxicity in patients who have received such drugs. Phototoxicity assessment is a regulatory requirement globally and one of the main toxicity screening steps in the early stages of drug discovery. An in silico-in vitro approach has been utilized mainly for toxicology assessments at these stages. Although several quantitative structure-activity relationship (QSAR) models for phototoxicity have been developed, in silico technology to evaluate phototoxicity has not been well established. In this study, we attempted to develop an artificial intelligence (AI) model to predict the in vitro Neutral Red Uptake Phototoxicity Test results from a chemical structure and its derived information. To accomplish this, we utilized an open-source software library, kMoL. kMoL employs a graph convolutional neural networks (GCN) approach, which allows it to learn the data for the specified chemical structure. kMoL also utilizes the integrated gradient (IG) method, enabling it to visually display the substructures contributing to any positive results. To construct this AI model, we used only the chemical structure as a basis, then added the descriptors and the HOMO-LUMO gap, which was obtained from quantum chemical calculations. As a result, the assortment of chemical structures and the HOMO-LUMO gap produced an AI model with high discrimination performance, and an F1 score of 0.857. Additionally, our AI model could visualize the substructures involved in phototoxicity using the IG method. Our AI model can be applied as a toxicity screening method and could enhance productivity in drug development.
INTRODUCTION
Phototoxicity has been defined by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use Safety 10 (ICH-S10) guideline as “an acute light-induced tissue response to a photoreactive chemical” (ICH-S10, 2013). It is possible that the phototoxicity of pharmaceutical products may induce serious adverse drug reactions in humans. Photosafety assessment data must, therefore, be submitted to the regulatory authority when filing applications for the approval of pharmaceuticals. As phototoxicity depends on optical and photophysical properties, a photosafety assessment should be conducted for chemicals with a Molar Extinction Coefficient (MEC) greater than 1000 L mol-1 cm-1 at wavelengths between 290 and 700 nm. The ICH-S10 guideline recommends conducting the 3T3 Neutral Red Uptake Phototoxicity Test (3T3 NRU-PT) in vitro before in vivo toxicity studies. If 3T3 NRU-PT indicates no phototoxicity potential, no further in vivo studies are required. The 3T3 NRU-PT is a widely used in vitro toxicology assay in the pharmaceutical industry (Bauer et al., 2021), and the test methods are standardized according to the Organization for Economic Co-operation and Development (OECD) guideline TG432 (OECD, 2019). In 3T3 NRU-PT, the uptake of neutral red in cultured mouse fibroblast cells, Balb/c 3T3, is used as an index, and phototoxicity is evaluated by comparing cytotoxicity with or without light irradiation. The 3T3 NRU-PT is conducted as both a Good Laboratory Practice (GLP) toxicology study, in the drug development stages, and a non-GLP toxicology screening assay, in the drug discovery stages. An in silico approach has been utilized for toxicology assessments in the early stages of drug development, and several QSAR models to predict phototoxicity has already been published (Ringeissen et al., 2011; de Lima Ribeiro and Ferreira, 2005). However, the development of a new in silico method for predicting phototoxicity with improved prediction accuracy would be indispensable. Consequently, we attempted to establish a phototoxicity prediction model using a deep learning method, the graph convolutional networks (GCN).
The rapid progress of AI technologies based on deep learning in recent years has been making a significant impact on the field of toxicology. In particular, GCN (Kipf and Welling, 2016), which learn the latent vectors of nodes and graphs, enable learning graph structures with practical computational complexity using only convolution. By considering the chemical structures as graphs, the GCN can learn the chemical structures themselves without converting the structure to other formats, such as descriptors. Furthermore, recent research, for example, via the integrated gradient (IG) method (Sundararajan et al., 2017), has resolved the problem of visualizing the AI decision-making process, that is, the basis for predictions, which has been considered unobservable until recently. A software package, kMoL (https://github.com/elix-tech/kmol), in which these functions were implemented, was developed by Kojima and Okuno from Kyoto University and Elix Inc. The kMoL software was developed based on kGCN (Kojima et al., 2020), which was constructed previously. The kMoL is a convolutional neural network framework implemented using the PyTorch library to create discriminant and regression models. It also includes features such as multimodal, multitasking, and federated learning (Kairouz et al., 2021).
As in the case of in vitro testing being used as an alternative to in vivo testing, in silico evaluation is expected to become the third experimental evaluation method, after in vivo and in vitro testing. Additionally, the in silico approach is an alternative when in vivo or in vitro studies cannot be reasonably performed for ethical, economic, or statistical reasons. In addition, in silico evaluation can be used to determine the experimental conditions for in vitro and in vivo evaluations. Despite these advantages, the ICH-S10 guidelines do not specify a specific in silico method for assessing phototoxicity. Thus, to improve the efficiency and increase the success rate of drug development, there is an urgent need to establish an in silico technology for predicting phototoxicity with high accuracy.
Conventional in silico methods to predict phototoxicity require chemical structures in in silico evaluation methods to be replaced with molecular descriptors and then learned using those descriptors. In contrast, GCN can learn the structure itself without any descriptors, which is, by simply considering the chemical structure as a graph structure without selecting the type of descriptor. This advantage eliminates any bias caused by the selection of molecular descriptors and allows the chemical structure to be directly learned.
Another approach to infer phototoxicity is to use the electronic structure information responsible for photoreactivity. The HOMO-LUMO gap, which is the energy difference between the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO), provides an estimate of the photoreactivity of chemicals. The smaller the gap, the more reactive the chemical. This quantity can be easily obtained from quantum chemical calculations. It has been used as a predictor of phototoxicity, along with various other descriptors for more than 15 years (de Lima Ribeiro and Ferreira, 2005; Haranosono et al., 2014; Schmidt et al., 2019).
In this research, by using kMoL employing GCN and an in vitro phototoxicity dataset, we created an AI model that simultaneously trained the graph structure and HOMO-LUMO gap calculated for each chemical structure to predict the result of in vitro phototoxicity test. The combination of descriptors was also examined. In addition, we visualized the chemical structures with contribution degrees that are considered to be involved in phototoxicity by employing the IG method. In this paper, we present research results and discuss the advantages and expected improvements of our AI model.
MATERIALS AND METHODS
Data preparation
In vitro phototoxicity datasets and the chemical structures encoded in the simplified molecular-input line-entry system (SMILES) were obtained from the data collected by Schmidt et al. (2019). They collected 3T3 NRU-PT information from the literature. Among these, 190 positive and 225 negative chemicals were obtained. For ex17a and ex17b in the data, the number of N bonds in the 5-membered ring in SMILES was four, which was corrected to three, as in an original paper by Hamaguchi et al. (2015). The modified SMILES were Cn1cc(-c2ccncc2)c(OCc2ccc(-c3nc4ccccc4n3C)cc2)n1 and Cn1cc(-c2ccncc2)c(OCc2ccc(-c3nc4ccc4n3C)cc2)n1, respectively. The photoirritation factor (PIF) used the data reported by Schmidt et al. (2019). Similar to Schmidt et al. when multiple values were available, the average was calculated and applied.
Molecular descriptors
One thousand six hundred thirteen descriptors generated from SMILES by 2D descriptors of Mordred (Moriwaki et al., 2018) version 1.2.0 were used as molecular-level features to input into the kMoL system.
Quantum chemical calculations
The chemical structure was initialized with ETKDGv3 (Riniker and Landrum, 2015) in RDKit, minimized with the Merck Molecular Force Field and then optimized at the B3LYP/6-31G* level using open-source quantum chemistry software Psi4 version 1.5 (Smith et al., 2020). Default convergence criteria (1.00 x 10-6 and 3.00 x 10-4 for energy and force, respectively) were used. The LanL2DZ basis set was used for chemicals involving iodine atoms to account for their relativistic effects. The HOMO-LUMO gap was calculated for the optimized structure at the same level. Three hundred ninety-nine structures were considered after excluding those that failed to converge in geometry optimization.
AI model construction
The AI model was first constructed using a GCN approach using only the chemical structures. Subsequently, we constructed AI models using a multi-modal GCN approach using descriptors, HOMO-LUMO gap values, or both, in addition to chemical structures. kMoL (https://github.com/elix-tech/kmol) was used to construct the AI models. The training, validation, and test sets were partitioned in the ratio of 8:1:1 using random sampling. The hyperparameter tuning was conducted using the training and validation sets with 5-fold cross-validation. The model performance was evaluated using the test set. A hyperparameter search was performed using the tree-structured Parzen estimator (TPE) algorithm implemented in Optuna (Akiba et al., 2019), and AdaBelief (Zhuang et al., 2020) was used for optimization. Mordred (Moriwaki et al., 2018) was used for descriptors. The metrics used in the present study are shown as follows.

The area under the receiver operating characteristic curve (ROC-AUC) and the area under the precision-recall curve (PR-AUC) are metrics used to evaluate models when the threshold is varied, using trade-off relationships between the true positive rate (recall) and false positive rate, in ROC-AUC, and between precision and recall, in PR-AUC.
RESULTS
The chemical structures encoded in SMILES and labels from the 3T3 NRU-PT results were used to train the AI model. The chemical structures were transformed into 45 features as molecular graph structures composed of nodes and edges (https://github.com/awslabs/dgl-lifesci/blob/master/python/dgllife/utils/featurizers.py). The hyperparameters for the AI were determined on the validation set, and the remaining data were used as a test set to evaluate the obtained model. An F1 score of 0.667 was obtained for the chemical structure alone. To improve this value, we added descriptors to the chemical structure and simultaneously recalculated the convolution, which resulted in only a slight improvement with an F1 score of 0.761 (Table 1).
Table 1. Performances of the models with each data set.
|
F1 |
Precision |
Recall |
Accuracy |
PR-AUC |
ROC-AUC |
| PIV |
0.667 |
0.786 |
0.579 |
0.756 |
0.810 |
0.802 |
| PIV + Mordred |
0.761 |
0.696 |
0.842 |
0.778 |
0.904 |
0.905 |
| PIV*1 + Egap*2 |
0.857 |
0.789 |
0.938 |
0.875 |
0.822 |
0.906 |
| PIV*1 + Egap*2 + Mordred |
0.788 |
0.765 |
0.813 |
0.825 |
0.762 |
0.878 |
| Schmidt model (random decision forests) |
0.88 |
0.86 |
0.90 |
0.85 |
n/a |
n/a |
The table shows the results of the holdout validation using the test set.
*1 PIV: the number of chemicals was reduced to 399, *2 Egap: HOMO-LUMO gap, n/a: not available.
Abbreviations: PIV; Phototoxicity in vitro data, Egap; HOMO-LUMO gap energy, PR-AUC; Area under the precision-recall curve, ROC-AUC; Area under the receiver operating characteristic curve.
Previous reports have shown that the HOMO-LUMO gap is useful for predicting phototoxicity. Therefore, to use this parameter to construct the AI model, the HOMO-LUMO gap was calculated for each chemical. Using the calculated HOMO-LUMO gap values for the 399 chemicals, which convolved with the above-mentioned 45 features obtained from the chemical structures, and an F1 score of 0.857 was obtained (Table 1).
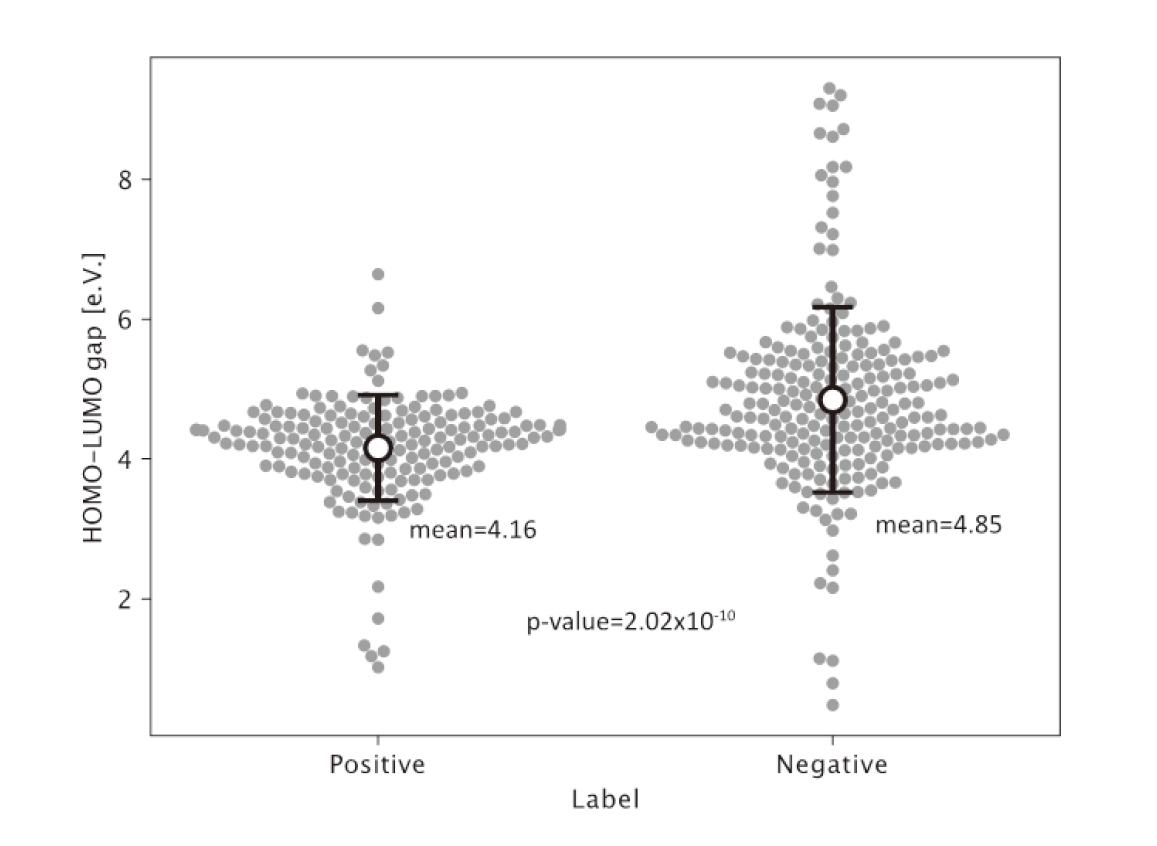
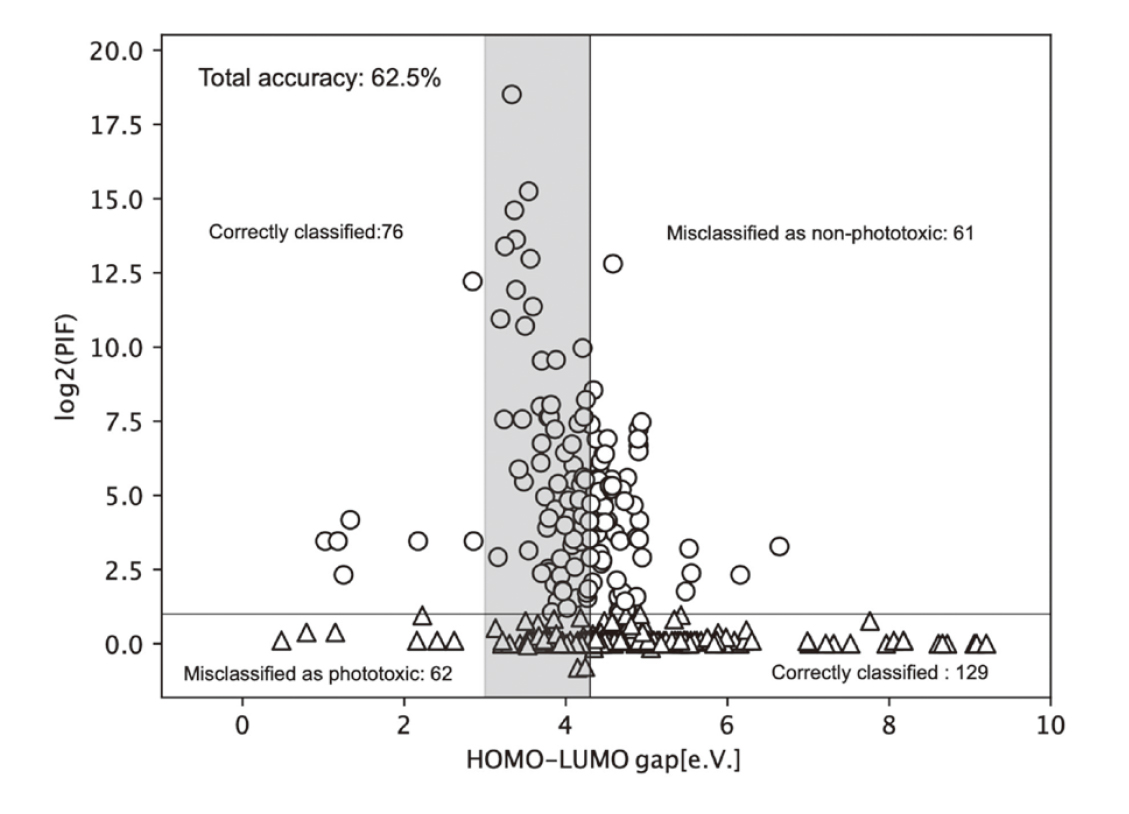
For further validation of our results, we examined whether it was possible to discriminate the two groups solely using HOMO-LUMO gap values. Schmidt et al. had a total accuracy of 59% for classification using the HOMO-LUMO gap alone with the semiempirical AM1 method (AM1); our total accuracy for classification with the density functional theory-based B3LYP method (B3LYP) was 62.5%. Thus, neither HOMO-LUMO gap alone resulted in an effective performance in classifying PIF values (Fig. 1). Although a statistically significant difference (p = 2.02 x 10-10 using Welch’s t-test) was observed between the positive and negative groups, it was not enough to distinguish which group the individual chemicals belonged to (Fig. 2).
We then aimed to further improve the model by adding the HOMO-LUMO gap to the chemical structure and descriptors. The 399 chemicals were modeled again, with 45 features obtained from the chemical structure and 1613 descriptors. The prediction accuracy did not improve, as expected (F1 score = 0.788) (Table 1). The combination of chemical structure information and HOMO-LUMO gap showed the best discrimination performance in this study.
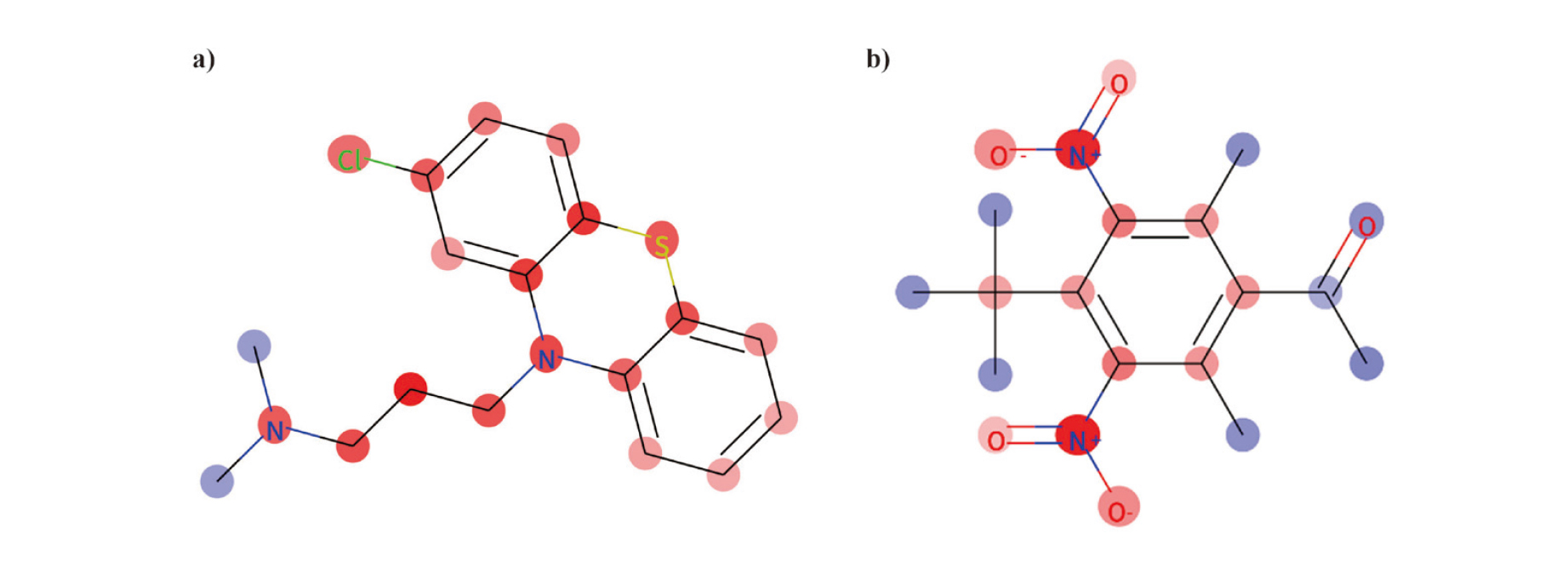
This AI model can visualize the structures that contribute to the outcomes using the IG method (Sundararajan et al., 2017). Examples of chlorpromazine and musk ketone are shown in Figs. 3a and 3b. In the present analysis, the phenothiazine backbone, including the chlorine atom and part of the piperazine-like ring side chain for chlorpromazine, and the nitro group for musk ketone, were correctly predicted as substructures that contributed to the predicted positive outcomes, respectively.
DISCUSSION
In this study, we constructed a phototoxicity prediction AI model using the kMoL and in vitro phototoxicity (3T3 NRU-PT) datasets. We confirmed that our AI model could predict phototoxicity with a high discrimination performance using fewer parameters than the prediction model by Schmidt et al.; furthermore, it could visualize the atomic groups contributing to the phototoxicity. The HOMO-LUMO gap is a parameter strongly related to the phototoxicity mechanism. The AI model was trained with kMoL using both the graph structure of the chemicals and the HOMO-LUMO gaps simultaneously. The discrimination performance (F1 score of 0.857) is comparable to the F1 score of 0.88 calculated by Schmidt et al. (2019) using the random decision forest approach and a set of 224 descriptors. They calculated and used 191 pharmacophoric fingerprints, nine HOMO-LUMO gaps, twenty-two spectral integrals, one ionization potential, and one electron affinity to construct their model. On the other hand, we achieved nearly comparable performance with only one HOMO-LUMO gap value and 45 features based on the graph structure derived from the chemical structure.
For further validation of our results, we examined whether it was possible to discriminate the groups solely using HOMO-LUMO gap values.
The approach Schmidt et al. took to classify PIF values using only the HOMO-LUMO gap did not yield useful results. This could have resulted from calculating the HOMO-LUMO gap values using AM1. The density functional theory-based B3LYP method calculates a more accurate electronic structure than the semiempirical approach for the AM1 generic function (Deeb and Clare, 2008). Therefore, we tried to classify the PIF values and construct the prediction model using the HOMO-LUMO gap values calculated with the more accurate B3LYP method. As a result, with B3LYP, the HOMO-LUMO gaps of the compounds were found distributed around an energy window of approximately 3.0–4.3 eV (290–400 nm), which is the generally expected UV transition energy region (Fig. 1). In contrast, AM1 yielded a window of 6.50–8.60 eV (Schmidt et al., 2019). The classification using the HOMO-LUMO gap and the PIF values resulted in an accuracy value of 62.5%, shown in Fig. 1. Although the accuracy is slightly better than the 59% of AM1, neither HOMO-LUMO gap alone performed well in classifying PIF values and predicting phototoxicity. Thus, a more accurate predictive performance can be achieved only when the HOMO-LUMO gap is combined with the information obtained from the chemical structure.
The F1 score for the AI model prediction was 0.667 when only structural information was used, and adding descriptors to the structural information only slightly improved the F1 score to 0.761. However, further addition of the HOMO-LUMO gap to the structural information and descriptors did not improve the prediction accuracy (F1 score of 0.788). This suggests that a better AI model can be constructed by adding only information related to the toxicity mechanism to the chemical structure. Our research results show that a multi-modal approach is effective in constructing AI models based on a GCN, and that the quality and optimization of parameters to be combined are more important than their number.
Our AI model successfully visualized the substructures involved in phototoxicity using the IG method. The performance can be viewed as an advantage over that of the prediction model constructed by Schmidt et al. IG is an interpretability and explainability technique for deep neural networks that visualizes the importance of the input features contributing to model predictions. Chlorpromazine has multiple photolysis pathways (Trautwein and Kummerer, 2012). Among the photolytic metabolites, dechlorinated chlorpromazine, in which the chlorine atom of the aromatic ring is replaced by a hydroxyl group, is reported to be highly reactive (Motten et al., 1985). Our IG visualization suggests that the phenothiazine scaffold, including chloride, contributes to phototoxicity. Additionally, in other words, kMoL can display the intensity of the positive and negative contributions to the predicted chemical. In our analysis, the nitro group of a musk ketone was predicted as a substructure that contributed to a positive outcome. In musk ketones, the photoreaction is initiated by an electronically excited nitro group intramolecularly attacking an adjacent tert-butyl group. This produces the intermediate indolenin-1-oxide, which is further converted to a ring structure (Zhao and Schwack, 2000).
Generally, the toxicological reactivities of the substructural alert can be influenced by other groups in the molecule because the chemical properties of the substructures depend on them (Alves et al., 2016). Therefore, the GCN approach, which can consider the entire structure, is suitable for identifying toxicity-relevant substructures and constructing AI models to predict toxicity from chemical structures. However, it is necessary to consider that kMoL analyzes two-dimensional (2D) chemical structures. Three-dimensional chemical structural information is required to analyze the HOMO-LUMO gap. Consequently, because it is difficult to construct an AI model in the current kMoL that also includes the HOMO-LUMO gap element, the HOMO-LUMO gap value must be calculated separately and loaded into the kMoL system. In addition, the current kMoL was not implemented to calculate the contribution degrees of the HOMO-LUMO gap value to the prediction results. If kMoL can be improved in the future to visualize the contribution degrees of the HOMO-LUMO gap, we will be able to obtain more information on the modification of chemical structures to avoid phototoxicity.
Here, we discuss the scope of application of our AI model. The 3T3 NRU-PT assay is an in vitro toxicology assay widely used in the pharmaceutical industry, and the test method has been standardized. For this reason, we used it as label information in the construction of the AI model. 3T3 NRU-PT is a method to evaluate acute light-induced tissue responses to photoreactive chemicals. Photoallergy, photogenotoxicity, and photocarcinogenicity are not subject to photosafety evaluation by the 3T3 NRU-PT. We did not use any other data, including clinical data, other than the 3T3 NRU-PT, in the AI model. Therefore, our AI model is positioned as a predictive model for acute phototoxicity assessment results in 3T3 NRU-PT. It should be noted that the 3T3 NRU-PT has a problem in that it is difficult to evaluate poorly soluble chemicals. Our AI model is expected to compensate for the weaknesses of the 3T3 NRU-PT.
Our AI model was developed as part of a collaborative industry-academia research. This project aimed to develop next-generation AI systems that contribute to research in the preliminary stages of drug discovery by applying AI technology, which has grown remarkably in recent years. The implemented AI systems are expected to contribute to the efficiency of drug development. The AI model constructed in this study was incorporated into the project’s AI system and operated.
We constructed an AI model to predict in vitro phototoxicity with a high discrimination performance. Simultaneously, this AI model can visualize the substructures involved in phototoxicity. This AI model has two features that simultaneously perform phototoxicity prediction and visualization of structural alerts: many prediction models reported thus far do not have these features, and these are believed to contribute to the improvement of chemical structures to avoid phototoxicity. Additionally, we demonstrated that the GCN approach, which we adopted for constructing AI models, is a useful method for developing toxicity prediction models. In the future, we expect to construct a wide variety of toxicity prediction models using the GCN approach.
ACKNOWLEDGMENT
This research was supported by the Japan Agency for Medical Research and Development (AMED) under Grant Number JP22nk0101111.
Conflict of interest
The authors declare that there is no conflict of interest.
REFERENCES
- Akiba, T., Sano, S., Yanase, T., Ohta, T. and Koyama, M. (2019): Optuna: A next-generation hyperparameter optimization framework. In: Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining), pp. 2623-2631.
- Alves, V., Muratov, E., Capuzzi, S., Politi, R., Low, Y., Braga, R., Zakharov, A.V., Sedykh, A., Mokshyna, E., Farag, S., Andrade, C., Kuz’min, V., Fourches, D. and Tropsha, A. (2016): Alarms about structural alerts. Green Chem., 18, 4348-4360.
- Bauer, D., Buckley, L.A., Delafoy, L., Ellinger-Ziegelbauer, H., Fellows, M.D., Gerets, H.H., Howe, J., Kaijser, G., Nicolette, J., Pettersen, B.A. and Schimpf, B. (2021): A cross-industry survey on photosafety evaluation of pharmaceuticals after implementation of ICH S10. Regul. Toxicol. Pharmacol., 125, 105017.
- de Lima Ribeiro, F.A. and Ferreira, M.M. (2005): QSAR model of the phototoxicity of polycyclic aromatic hydrocarbons. J. Mol. Struct. THEOCHEM, 719, 191-200.
- Deeb, O. and Clare, B.W. (2008): Comparison of AM1 and B3LYP-DFT for inhibition of MAO-A by phenylisopropylamines: a QSAR study. Chem. Biol. Drug Des., 71, 352-362.
- Hamaguchi, W., Masuda, N., Miyamoto, S., Kikuchi, S., Narazaki, F., Shiina, Y., Seo, R., Amano, Y., Mihara, T., Moriguchi, H. and Hattori, K. (2015): Addressing phototoxicity observed in a novel series of biaryl derivatives: discovery of potent, selective and orally active phosphodiesterase 10A inhibitor ASP9436. Bioorg. Med. Chem., 23, 3351-3367.
- Haranosono, Y., Kurata, M. and Sakaki, H. (2014): Establishment of an in silico phototoxicity prediction method by combining descriptors related to photo-absorption and photo-reaction. J. Toxicol. Sci., 39, 655-664.
- ICH-S10. (2013): ICH S10 Photosafety evaluation of pharmaceuticals), https://www.ema.europa.eu/en/ich-s10-photosafety-evaluation-pharmaceuticals (accessed 27 September 2022).
- Kairouz, P., McMahan, H.B., Avent, B., Bellet, A., Bennis, M., Bhagoji, A.N., Bonawitz, K., Charles, Z., Cormode, G., Cummings, R., D’Oliveira, R.G.L., Eichner, H., Rouayheb, S.E., Evans, D., Gardner, J., Garrett, Z., Gascón, A., Ghazi, B., Gibbons, P.B., Gruteser, M., Harchaoui, Z., He, C., He, L., Huo, Z., Hutchinson, B., Hsu, J., Jaggi, M., Javidi, T., Joshi, G., Khodak, M., Konecný, J., Korolova, A., Koushanfar, F., Koyejo, S., Lepoint, T., Liu, Y., Mittal, P., Mohri, M., Nock, R., Özgür, A., Pagh, R., Qi, H., Ramage, D., Raskar, R., Raykova, M., Song, D., Song, W., Stich, S.U., Sun, Z., Suresh, A.T., Tramèr, F., Vepakomma, P., Wang, J., Xiong, L., Xu, Z., Yang, Q., Yu, F.X., Yu, H. and Zhao, S. (2021): Advances and open problems in federated learning. Foundations and Trends® in Machine Learning, 14, 1-210.
- Kipf, T.N. and Welling, M. (2016): Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907.
- Kojima, R., Ishida, S., Ohta, M., Iwata, H., Honma, T. and Okuno, Y. (2020): kGCN: a graph-based deep learning framework for chemical structures. J. Cheminform., 12, 32.
- Moriwaki, H., Tian, Y.S., Kawashita, N. and Takagi, T. (2018): Mordred: a molecular descriptor calculator. J. Cheminform., 10, 4.
- Motten, A.G., Buettner, G.R. and Chignell, C.F. (1985): Spectroscopic studies of cutaneous photosensitizing agents--VIII. A spin-trapping study of light induced free radicals from chlorpromazine and promazine. Photochem. Photobiol., 42, 9-15.
- OECD. (2019): Test No. 432: In Vitro 3T3 NRU Phototoxicity Test.), https://www.oecd-ilibrary.org/content/publication/9789264071162-en (accessed 27 September 2022).
- Ringeissen, S., Marrot, L., Note, R., Labarussiat, A., Imbert, S., Todorov, M., Mekenyan, O. and Meunier, J.R. (2011): Development of a mechanistic SAR model for the detection of phototoxic chemicals and use in an integrated testing strategy. Toxicol. In Vitro, 25, 324-334.
- Riniker, S. and Landrum, G.A. (2015): Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Model., 55, 2562-2574.
- Schmidt, F., Wenzel, J., Halland, N., Güssregen, S., Delafoy, L. and Czich, A. (2019): Computational Investigation of Drug Phototoxicity: Photosafety Assessment, Photo-Toxophore Identification, and Machine Learning. Chem. Res. Toxicol., 32, 2338-2352.
- Smith, D.G., Burns, L.A., Simmonett, A.C., Parrish, R.M., Schieber, M.C., Galvelis, R., Kraus, P., Kruse, H., Di Remigio, R., Alenaizan, A., James, A.M., Lehtola, S., Misiewicz, J.P., Scheurer, M., Shaw, R.A., Schriber, J.B., Xie, Y., Glick, Z.L., Sirianni, D.A., O’Brien, J.S., Waldrop, J.M., Kumar, A., Hohenstein, E.G., Pritchard, B.P., Brooks, B.R., Schaefer, H.F. 3rd, Sokolov, A.Y., Patkowski, K., DePrince, A.E. 3rd, Bozkaya, U., King, R.A., Evangelista, F.A., Turney, J.M., Crawford, T.D. and Sherrill, C.D. (2020): Psi4 1.4: open-source software for high-throughput quantum chemistry. J. Chem. Phys., 152, 184108.
- Sundararajan, M., Taly, A. and Yan, Q. (2017): Axiomatic attribution for deep networks. In: International conference on machine learning), pp. 3319-3328, PMLR.
- Trautwein, C. and Kümmerer, K. (2012): Degradation of the tricyclic antipsychotic drug chlorpromazine under environmental conditions, identification of its main aquatic biotic and abiotic transformation products by LC-MSn and their effects on environmental bacteria. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., 889-890, 24-38.
- Zhao, X. and Schwack, W. (2000): Photochemical reactions of musk ketone. Toxicol. Environ. Chem., 74, 217-230.
- Zhuang, J., Tang, T., Ding, Y., Tatikonda, S.C., Dvornek, N., Papademetris, X. and Duncan, J. (2020): Adabelief optimizer: adapting stepsizes by the belief in observed gradients. Adv. Neural Inf. Process. Syst., 33, 18795-18806.